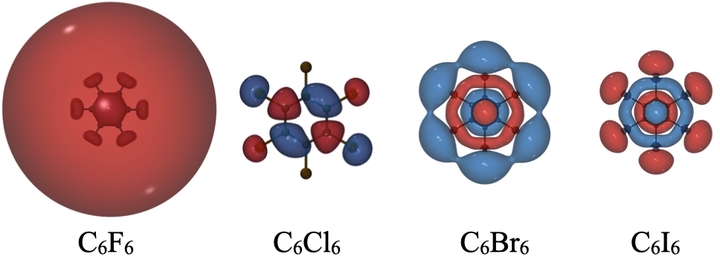
One of the crucial parameters that defines the performance of organic semiconducting materials is its charge carrier mobility. This intrinsic property is determined by temperature, crystallinity of the material, electronic and vibronic couplings. We are investigating new charge transport pathways due to diffuse electronic states that span several monomer units. These states may lead to larger electronic couplings and lower barriers to charge transfer. Our recent investigations have shown that such states are present in a variety of organic molecules ranging from halogenated aromatic systems to highly conjugated fullerene molecules. We are also investigating the role of underlying crystal structure in determining the electronic couplings.
Charge mobility in organic materials