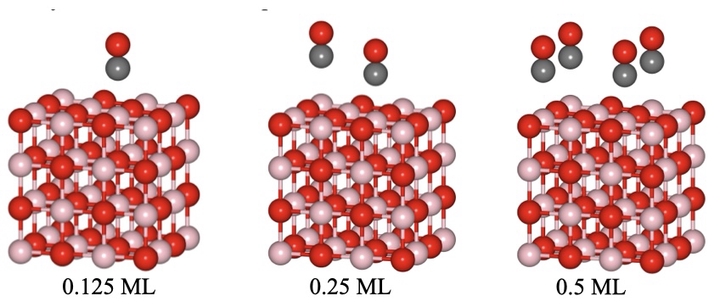
Molecular adsorption on material surfaces is the precursor to chemical transformations. However, traditional density functional approximations show puzzling errors in describing molecular adsorption. This critical drawback hampers reliable modeling of molecular reactions and separations via materials. We are therefore analyzing the sources of these puzzling errors while developing low-scaling computational solutions. Our recent studies show that density-corrected KS and many-body methods can overcome these errors leading to remarkable improvements.
Many-body methods for surface chemistry